

Contents
Overview
The M. tuberculosis TB-Profiler AMR Task Template allows to use whole genome sequencing data to predict lineage and drug-resistance for Mycobacterium tuberculosis complex samples [1]. It uses the tool TB-Profiler to process the FASTQ read files for the sample (without applying any downsampling or trimming). This tool aligns reads to the H37Rv reference using bwa for Illumina or minimap2 for ONT data and then calls variants using bcftools. These variants are then compared to a drug-resistance database. The tool also predicts mixed-strain infections and the number of reads supporting drug resistance variants as an insight into hetero-resistance (latter not applicable for ONT data; PacBio data are currently not at all supported).
Requirements
![]() Important:
Important:
- If Ridom Typer is installed on Windows, TB-Profiler requires the Windows Subsystem for Linux (WSL).
- If Ridom Typer is installed on Linux, TB-Profiler must once be installed by calling the installation of Bioinformatic Tools on Linux.
- The task template can be used with Illumina and ONT reads.
Database and Parameters
TB-Profiler uses the mutations from the 2nd edition WHO mutation catalogue and the TB-Profiler original library. The used database can be changed by editing the genotyping library (2nd edition WHO mutation catalogue only) in the task template editor.
TB-Profiler is run with default command line parameters that cannot be changed by the user:
--depth DEPTH Minimum depth hard and soft cutoff, default: 0, 10 --af AF Minimum allele frequency hard and soft cutoff, default: 0, 0.1 --strand STRAND Minimum read number per strand hard and soft cutoff, default: 0, 3 --sv_depth SV_DEPTH Structural variant minimum depth hard and soft cutoff, default: 0, 10 --sv_af SV_AF Structural variant minimum allele frequency hard cutoff, default: 0.5, 0.9 --sv_len SV_LEN Structural variant maximum size hard and soft cutoff, default: 100000, 50000
For ONT data, the additional CLI parameter --platform nanopore is used.
With these default parameters TB-Profiler will filter out mutations (applies also for hetero-resistance detection) which
- have a frequency between 0% and 10%
- are found on more than one and less than 10 reads in total
- are found on less than 3 reads in forward or reverse direction
and display them in the "QC failed" section with comment "soft fail". Results that do not match the hard cutoff are not displayed at all.
Task Entry Overview
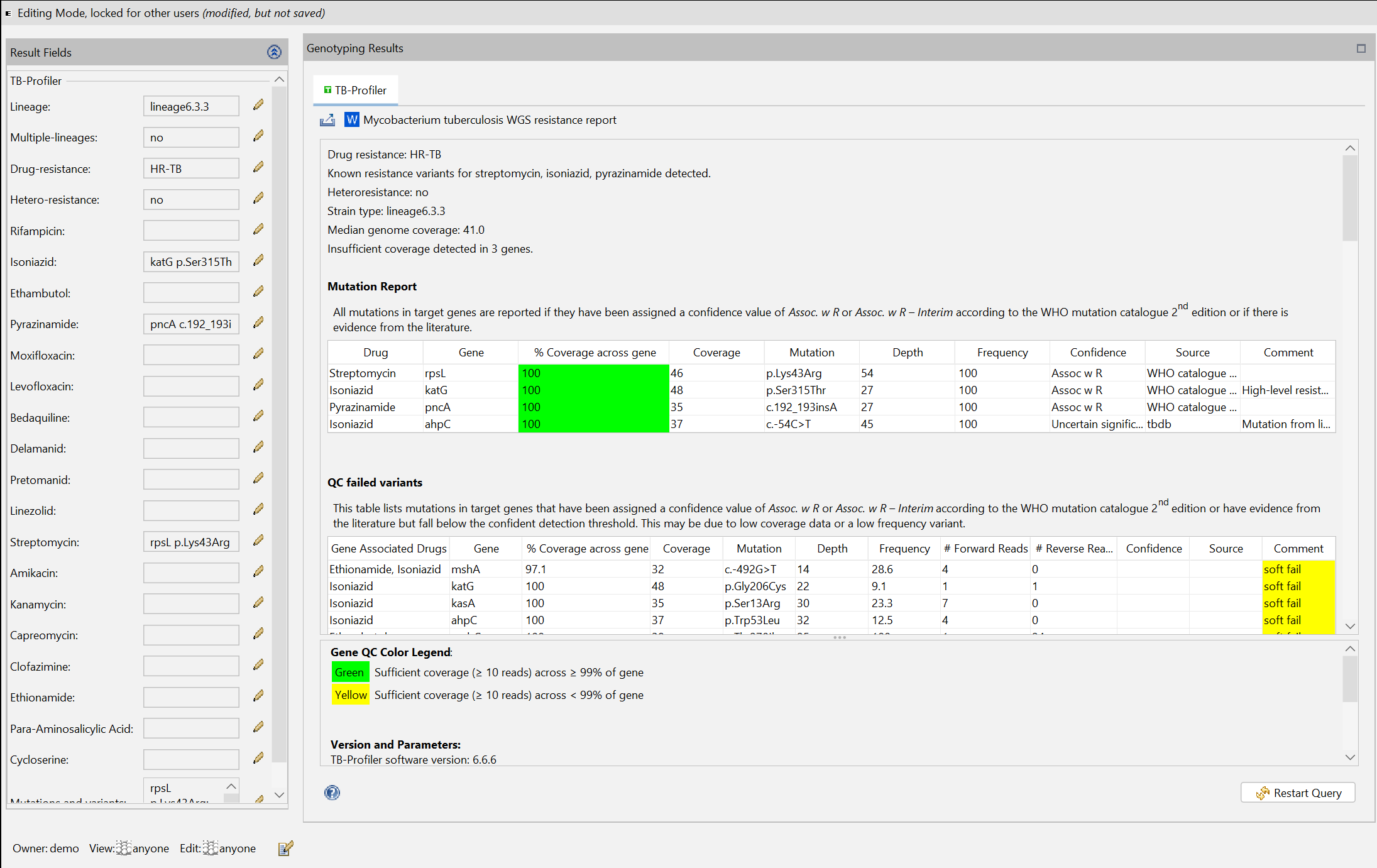
The task entry contains an overview that displays the TB-Profiler results. On top a MS Word file report can be opened using the ![]() Mycobacterium tuberculosis WGS resistance report button. The button
Mycobacterium tuberculosis WGS resistance report button. The button ![]() Export results allows to export the TB-Profiler results as a text-, MS Word-, or machine-readable .json-file.
Export results allows to export the TB-Profiler results as a text-, MS Word-, or machine-readable .json-file.
In the TB-Profiler results, the column % Coverage across gene is highlighted in green if all of the gene has sufficient coverage across ≥ 99% of gene and yellow otherwise. The minimum coverage required for sufficient coverage is 10 reads (see default parameters above).
The term Coverage refers to the median coverage across a gene.
The term Depth refers to the sequencing depth at a specific genome position. This is the number of individual reads that cover this location.
The term Frequency indicates how often a particular mutation was found in the sequence data that was read in. It therefore describes the proportion of reads that carry this mutation.
Drug resistance class definitions
Samples are classed into different types using the following definitions (see TB-Profiler manual):
| Type | Drugs resistance |
|---|---|
| Sensitive | No drug resistance |
| Pre-MDR | Rifampicin or isonisazid |
| MDR | Rifampicin and isoniazid |
| Pre-XDR | MDR and any fluoriquinolone |
| XDR | MDR and (any fluoriquinolone and any group A drug) |
| Other | Resistance to any drug but none of the above categories |
Result Fields
Several result fields are stored from the TB-Profiler output:
- Lineage, e.g. 4.7
- Multiple-lineages: is set to true if more than one lineage is reported
- Drug-resistance, e.g. Pre-XDR-TB
- Hetero-resistance: is set to true if the Mutation Report has a frequency ≠ 100 for a drug-resistance mutation
- Known resistance variants for rifampicin, isoniazid, ethambutol, pyrazinamide, moxifloxacin, levofloxacin, bedaquiline, delamanid, pretomanid, linezolid, streptomycin, amikacin, kanamycin, capreomycin, clofazimine, ethionamide, para-aminosalicylic acid, and cycloserine, e.g. Isoniazid katG p.Ser315Thr"
- Mutations: all mutations from the Mutation Report and Other Variants tables listed by TB-Profiler, e.g. katG p.Ser315Thr and/or gyrA c.-422G>A
Lineages are assigned by looking for lineage-specific SNPs. The initial list of SNPs was published by Coll et al. [2]. The SNP barcode was further refined by Napier et al. [3] and Zwyer et al. [4].
Multiple lineages may result from a mixed-strain infection (especially in high-incidence settings), (laboratory) contamination, or issues with low coverage. Multiple lineages are detected if the two strains differ at least in the first decimal digit of the lineages (nested multiple lineages, e.g., lineages 4.1.1 and 4.1.2, are usually not found). Each lineage must have support for defining SNPs with 2% or more reads and at least 5 reads in total.
Multiple lineages can but must not result in hetero-resistance and vice versa.
Searching for Results
The result fields can be used to search for a sample with a specific drug resistance and/or mutation.
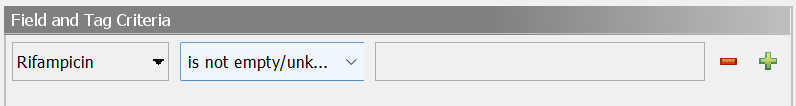
As an example, to search for samples with reported rifampicin resistance, Rifampicin is not empty/unknown can be used in the advanced search samples dialog:
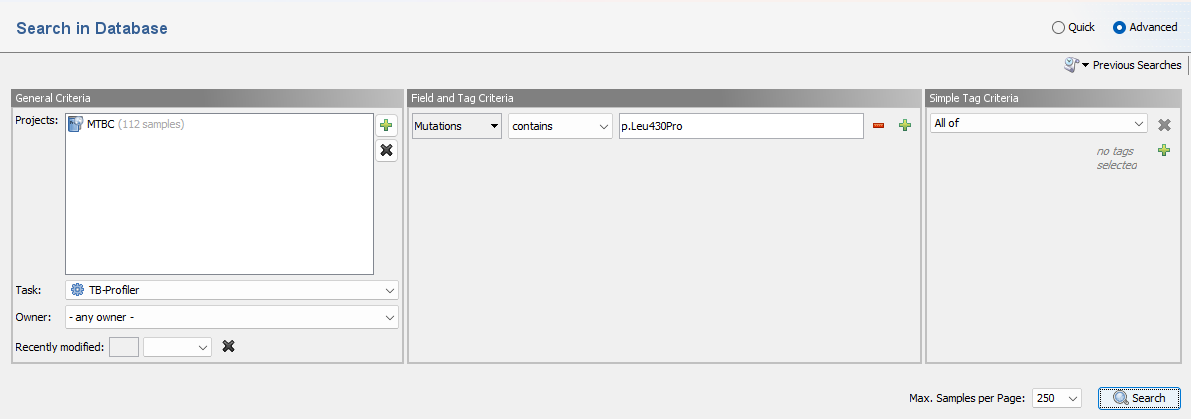
To search for a sample with a specific mutation use a query like Mutation contains p.Leu430Pro.
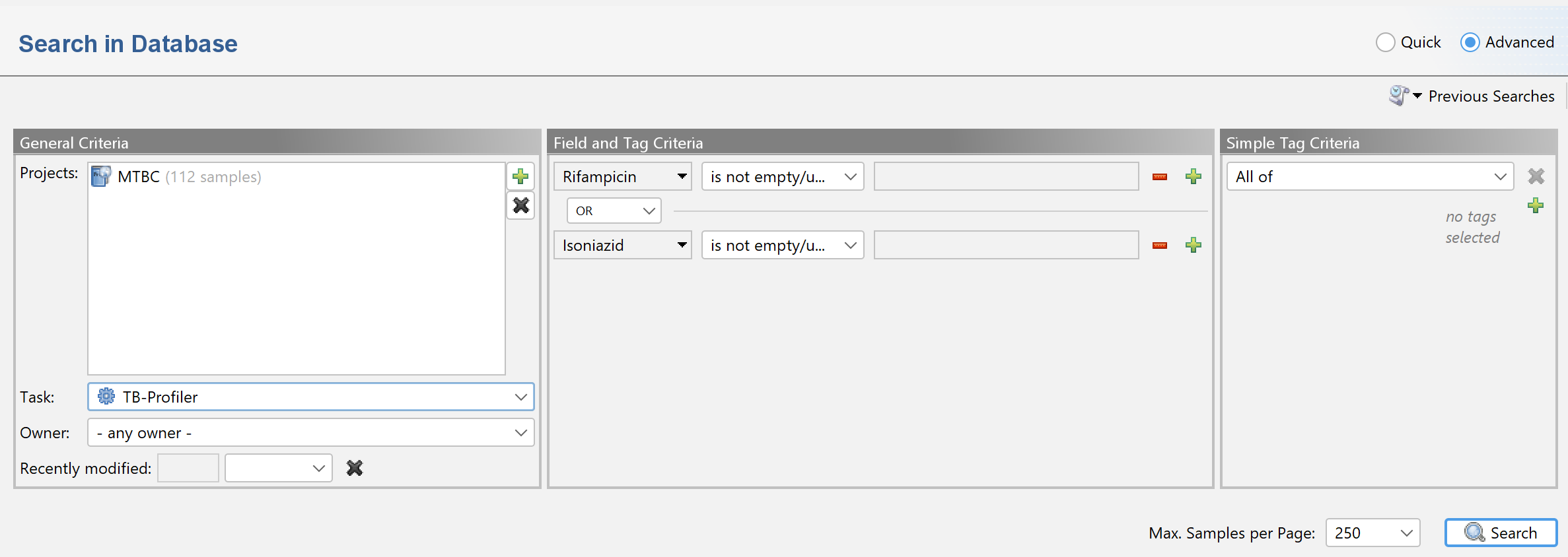
Note that search queries can be combinded by Boolean operators (e.g., AND or OR). For example, to search for samples with a reported resistance to rifampicin or isoniazid use
Rifampicin is not empty/unknown OR Isoniazid is not empty/unknown:
Batch Export
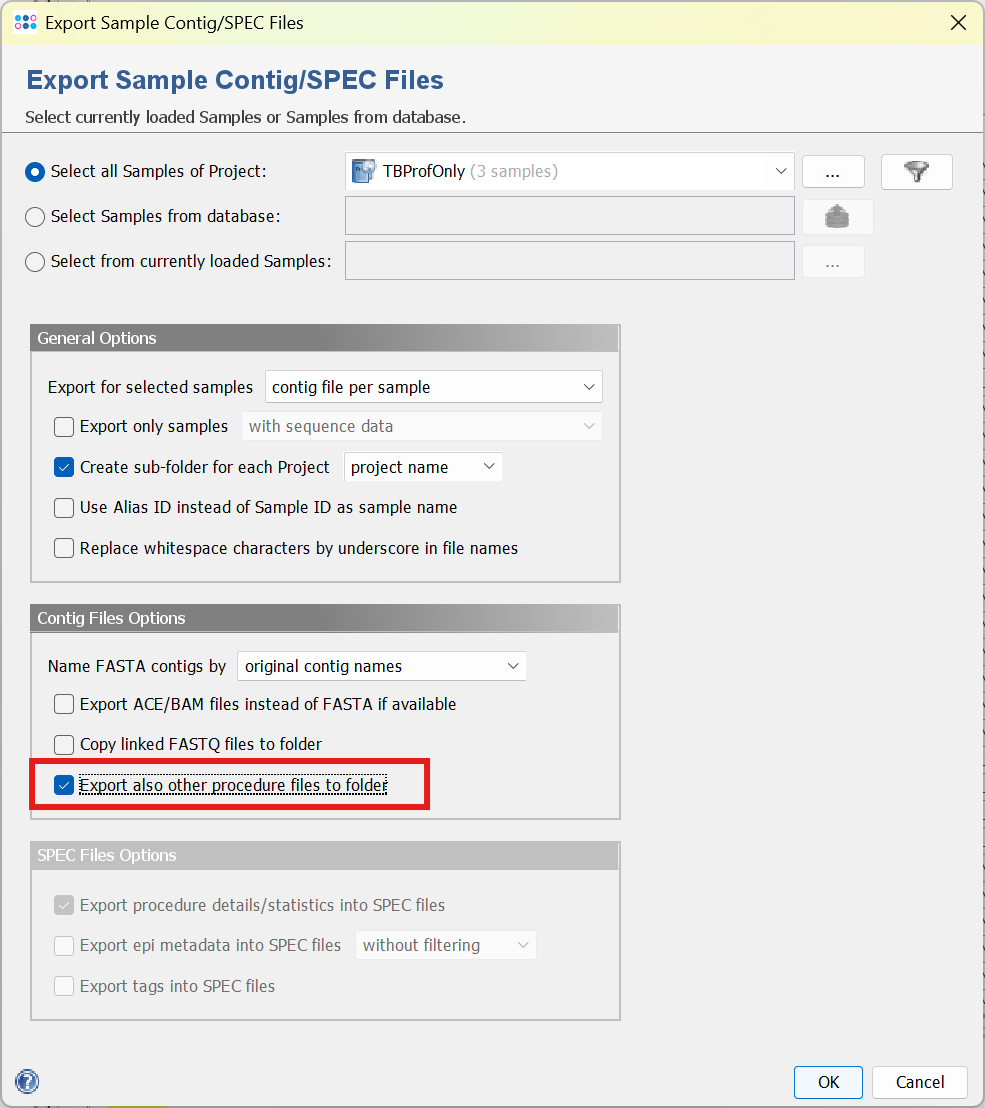
The results from TB-Profiler can be exported using the menu function File > Export Sample Contig/SPEC Files. Under Contig File Options select Export also other procedure files to folder. This will add the TB-Profiler Word-files and the .json files to the export.
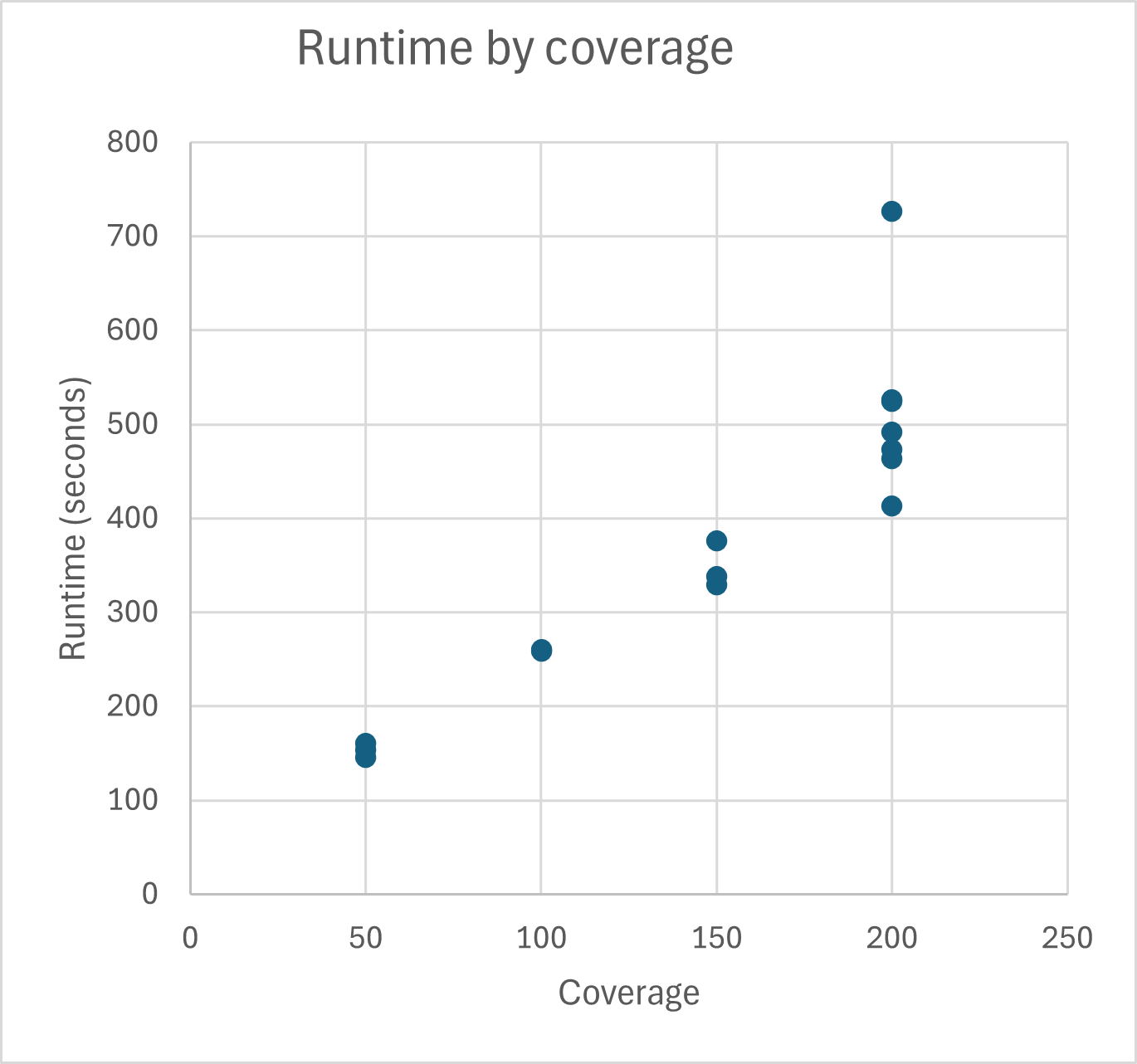
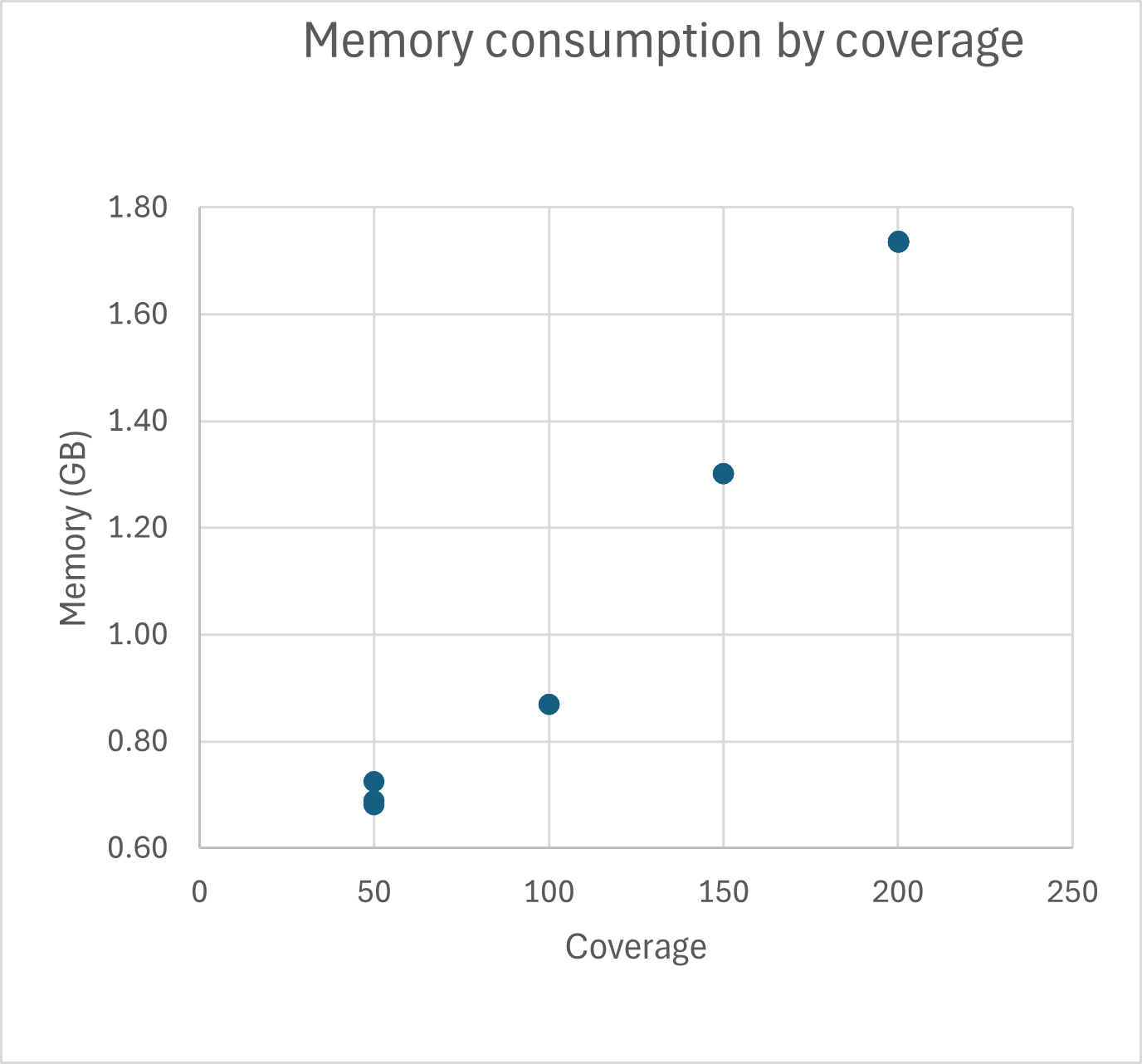
Runtime and memory consumption
TB-Profiler runtime and memory consumption was tested for Illumina 2x 250bp reads. Typical runtime for TB-Profiler for 200x coverage is around 500 seconds on an Intel Core i7-13850 system when 4 cores are used. Typical memory consumption for 200x coverage is around 1.7 GB when 4 cores are used.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.